
Researchers from the Sequencing Facility and Biomedical Informatics and Data Science Directorate at the Frederick National Laboratory (FNL) collaborated with the U.S. Food and Drug Administration (FDA) and other research centers across the U.S. to conduct a benchmark study establishing best practices for high-throughput single-cell RNA sequencing.
The study, published in Nature Biotechnology, provides guidance on selecting the ideal combination of technology platforms and bioinformatic methods based on experimental needs.
The single-cell sequencing boom
Sequencing enables researchers to identify the transcriptome or genome for all RNA or DNA, respectively, present in a sample. Before single-cell sequencing became available, scientists could do this through bulk sequencing.

A schematic detailing the design of the study, including the two cell lines, and following each step from start to finish. Figure first appeared in Nature Biotechnology.
In bulk sequencing, all the DNA or RNA in a sample (including all cell types) are analyzed together. Now, because of single-cell sequencing, researchers are able to do the same for each individual cell in a sample. This provides powerful information for researchers.
“You can look at cell populations within samples that are resistant to—or susceptible to—different therapies. Or subpopulations in tumors that can better metastasize—or don’t form metastatic tumors. It gives us much more in-depth information that can be really valuable when we are trying to understand the biology of normal and disease states,” said Monika Mehta, the research and development manager at FNL’s Sequencing Facility.
As a result, the technology has become increasingly popular in biomedical research. The scientists at FNL’s Sequencing Facility, for instance, tripled the volume of single-cell samples processed in just one year, going from processing around 880 single-cell sequencing samples in 2019 to more than 3,000 in 2020.
“It has exploded. … Everybody wants to and almost everybody is doing single-cell sequencing,” said Mehta.
But single-cell sequencing is still a relatively new method, having been around only for about a decade (and the high-throughput version used in FNL’s Sequencing Facility today, only a few years). Scientists are still fine-tuning how they work with it.
Building confidence in data
“Just like any new technology, things need to get standardized, and things will need to have a benchmark in order to compare the performance between facilities,” said Bao Tran, the Sequencing Facility’s laboratory director.
The technology itself is prone to high variability because cells in a sample can be at different stages of life or health when they’re processed. Cells begin to decline and die once they’re removed from a patient’s body. There may also be very different cell types within the same sample.
In bulk sequencing, this information “averages out” when scientists look at the most prominent DNA or RNA. But in single-cell sequencing, this variation matters a lot because it can change the interpretation of the data.
“It was important to see how much confidence we can have in the data,” said Mehta.
This need brought about the collaboration with researchers from the FDA and several other sequencing centers, under the Sequencing Quality Control Phase 2 consortium, to try to create a benchmark for quality control in single-cell RNA sequencing.
An experiment to help with experimental design
The FDA provided the same two cell lines (breast cancer and B lymphocyte cells) to each of the sequencing facilities to carry out single-cell library preparation and sequencing. This benchmark study used various combinations of preprocessing, normalization, batch correction, and analysis methods on the most common and popular sequencing platforms for either full-length RNA or shorter strands.
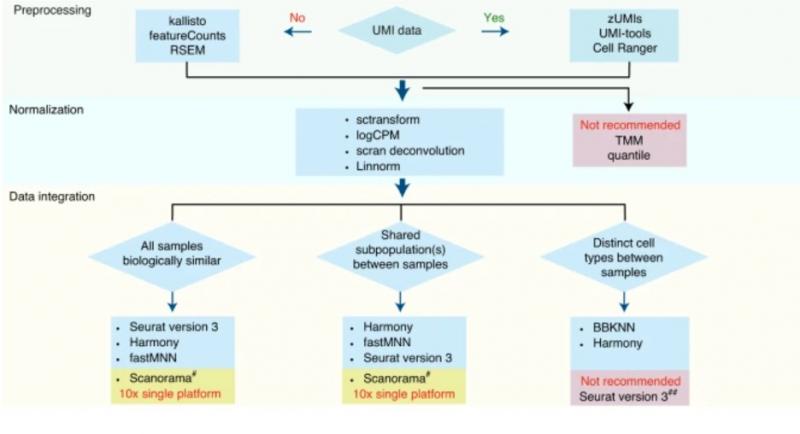
A flow chart, developed during the study, of best practice recommendations for single-cell RNA sequencing analysis. Figure first appeared in Nature Biotechnology.
FNL also tested two other variables. One was a method for fixing the samples before processing, which may prevent some changes to the cells during transport. The second tested samples of B lymphocytes with either 5% or 10% of cancer cells mixed in to see if the methods were sensitive enough to read those amounts of tumor cells. (Some were and some weren’t.)
The results demonstrated the importance of normalization and batch correction, especially for single-cell sequencing.
“When analyzing those two samples across the platforms, you can clearly see very different results. That platform effect is really dominant and even greater than the biological differences, so that’s why it’s important to apply software algorithms to be able to actually correct those batch effects,” said Yongmei Zhao, the bioinformatics manager of the Sequencing Facility’s bioinformatics group, also part of the Biomedical Informatics and Data Science Directorate.
“This study really highlights the importance of choosing the computational method appropriate both for the technology and based on the sample heterogeneity and cell subpopulation composition,” she added.
Because the researchers used two cell lines that are already well characterized, they knew what to expect in the results. This made it easier to know whether they were making the right adjustments before analysis and whether their final data were accurate.
“It’s kind of like trying all these different pairings to see … what impacts the results, and which one looks like it has the best output that matches what you would expect biologically. … It turns out, certain methods perform better on certain … types of data … so it makes analysis potentially a little more complicated since you’ll actually need to understand your data a little bit more before you know what’s the best way to go and approach certain things,” said Vicky Chen, an analyst on Zhao’s team.
Though it may make experimental design planning a little harder up front, the study provides valuable information. By taking the time to plan accordingly, researchers won’t waste time and resources to sequence something that may not yield accurate results.
Creating a ‘truth set’ of data
After analyzing their data, the groups determined the best combination of methods depending on sample type for the purposes of an experiment. They created a usable flow chart with the information.
Even if researchers want to use different software or platforms, the data generated in this study can still help them.
“It’s publicly available to the research community so they can also download the data that we generated to benchmark a newer platform … or new software. … So that is another benefit and also a resource that we, through this study, offer to the community,” said Zhao.
FNL’s Sequencing Facility will likewise benefit from this resource as they come across any new platforms to test.
“We now have sort of a ‘truth set’ available. Whenever we … test any new technologies, any new single-cell platforms, these are the two cell lines we use as a reference. … In effect, we are continuously building our resource every time we use [these cell lines],” said Mehta.
Continuous improvement
This study provided useful benchmark information to help with single-cell sequencing quality control, but the technology continues to rapidly evolve.
The group plans to do its part to make sure benchmarks will hold their value over time. Working toward best practices and continually learning from and sharing with the community is an integral part of their work.
“Single-cell technology is a very fast-moving target. … We will have to continue working on the next thing and establish, reestablish the benchmarks again in the near future,” said Tran.
Media Inquiries
Mary Ellen Hackett
Manager, Communications Office
301-401-8670